Son varias las parasitosis que ocurren en el torrente sanguíneo. Son
causadas por los protozoos que al menos
en una fase de su desarrollo transitan en la sangre, lo que se conoce como parasitemia. Las más
importantes en nuestro país son: las especies del género Plasmodiu, Trypanosoma
y Leshmania. ( Rodak,2004)
Malaria
o Paludismo.
Son los dos términos empleados para
definir una misma enfermedad producida por un parásito (protozoo) del género Plasmodium que es transmitido por la
picadura de las hembras del mosquito anófeles (Género Anopheles).
Existen cuatro especies de plasmodios
que pueden afectar al hombre: Plasmodium falciparum , Plasmodium vivax , Plasmodium malariae y Plasmodium ovale . De todos ellos la
infección más común es la causada por P. falciparum, que además es la que causa
una enfermedad más grave siendo la que
mayor número de muertes produce. (Malaria o paludismo, s.f).

(Malaria o
paludismo, s.f)
Plasmodium
falciparum.
Enfermedades causadas: Paludismo, infección hepática y de la sangre transmitida por mosquitos.
Patogenia: Los esporozoitos infectan las
glándulas salivales de mosquitos y se transmiten al ser humano a través de la
picadura, infectando hepatocitos y multiplicándose en esquizontes que se
convierten en merozoitos uninucleados que invaden hematíes, en los que el
merozoito madura en gametocito (estadio sexual) o en más merozoitos. Si los
gametocitos infectan al mosquito a través de la picadura, maduran en nuevos
esporozoitos en sus glándulas salivales, repitiéndose el ciclo. Los esquizontes
de los hematíes liberan más merozoitos que infectan a más hematíes. La fiebre y
escalofríos son consecuencia de la lisis de hematíes. El paludismo por P. falciparum puede obstruir los vasos sanguíneos
cerebrales (y otros órganos) dando lugar a complicaciones de gravedad.
Es el más
patógeno de los agentes del paludismo humano y la infección que provoca puede
tener una evolución fulminate aguda, que conduce a la muerte si el
tratamiento no se inicia de inmediato.
Es importante establecer un diagnostico temprano y rápido. (Rodak, 2004).

Figura 126.
Cuadro Morfológico
Frotis Fino:
1.
Eritrocitos no parasitados
2.
Trofozoitos
jóvenes
3.
Trofozoitos maduros
4.
Esquizontes Jóvenes
5.
Esquizontes maduros
6.
Macrogametocitos
7.
Microgametocitos.

Figura 127
Muestra: Trofozoitos.
Tincion: Giemsa: Aumento 1000x
Tincion: Giemsa: Aumento 1000x
Fuente.
(Rodak, 2004)
Plasmodium
Malariae.
Parásitos invaden los eritrocitos más
viejos y la infección suele ser
leve. El estadio anular tiene un solo
punto de cromatina y un anillo de
citoplasma de color azul que suele ser más chico y denso que el de P. vivax
pero no pueden distinguirse de él. El trofozoito en crecimiento la cromatina es
redondeada o estriada, el citoplasma adopta la forma de una banda estrecha que atraviesa la célula
y una característica de la especie. Los eritrocitos no aumentan de tamaño y no
hay granulaciones en el estadio de esquizonte.

Figura128.
Muestra: Esquizonte.
Tincion: Giemsa: Aumento 1000x
Fuente. (Malaria, s.f)

Figura 129. Muestra:
Morfologia.
Fuente: (Malaria, s.f)
PLASMODIUM VIVAX.
Tiene
una distribución mundial, sobre todo en los climas templados y en los trópicos. No es tan frecuente en
África como los otros tipos. Es difícil de erradicar y puede haber
recidivas. El esquizonte de P. vivax permanece latente durante años en el
hígado y puede reactivarse y provocar
una recidiva de la infección. (Rodak, 2004)

Figura 130.
Muestra: Trofozoito.
Tincion: Giemsa: Aumento 1000x
Tincion: Giemsa: Aumento 1000x
Fuente: (Malaria,
s.f)
MORFOLOGÍA
En el
ciclo eritrocítico se encuentran las siguientes formas: trofozoíto anular,
trofozoíto ameboide, esquizonte inmaduro, esquizonte maduro, macrogametocito y
microgametocito.
› Trofozoíto anular. Ocupa aproximadamente 1/3 del diámetro del eritrocito;
se observa un punto grande de cromatina y un circulo de citoplasma.
› Trofozoíto
ameboide. El trofozoíto anular incrementa el tamaño de la
cromatina y del citoplasma, algunas veces llena prácticamente el glóbulo rojo.
› Esquizonte
joven. La cromatina se divide en dos o más masas irregulares; el
citoplasma muestra varios grados de separación y el pigmento tiende a
compactarse hacia un lado del parasito.
› Trofozoíto
maduro. Usualmente presenta 16 merozoítos, cada uno con un gránulo de
cromatina y una pequeña masa circulas del citoplasma. El pigmento se aglomera
en uno o dos grupos. Algunas veces el parásito llena el glóbulo rojo.
› Macrogametocito. Es
redondeado u ovalado; el citoplasma es homogéneo y no presenta vacuolas. La
cromatina es pequeña, compacta y usualmente excéntrica. El pigmento malárico se
concentra en el citoplasma.
› Micromegatocito. Su
citoplasma es pequeño y generalmente la cromatina es difusa y central. El
pigmento malárico se encuentra disperso en el citoplasma. : P.vivax.
(2012, Junio 21).

Figura. 131.
Muetra: Cuadro morfológico.
Fuente: P.vivax.
(2012, Junio 21).
Plasmodium
Ovale.
Los parásitos invaden eritrocitos
jóvenes y reiculocitos. El trofozoíto joven se parece al de P.vivax y al
de P. malariae. Los eritrocitos aumentan
de tamaño una forma ovalada y crenada, y tiene gránulos de Schüffner. En el
estadio de esquizonte los eritrocitos son ovalados y el parásito es redondeo y
se ubica en el centro de la célula. Los merozoítos pueden ser de cuatro a ocho
o más y se encuentran en forma de roseta alrededor de una masa de pigmento. Los
gametocitos no pueden diferenciarse de los
P.vivax. La duración del ciclo asexual es de 48 horas. (Rodak, 2004).

Figura
132.
Cuadro Morfológico.
Fuente: (Malaria, s.f)
Cuadro Morfológico.
Fuente: (Malaria, s.f)

Figura 133. Esquizonte inmaduro Muestra.
Sangre periférica
Tinción: Giemsa. Aumento 1000x
Fuente: Malaria (s.f).
Tripanosomiasis
americana.
La tripanosomiasis americana ( enfermedad de
Chagas) está producida por el Trypanosoma cruzi, que es transmitido por el
chinche Reduviidae, subfamilia triatominae. La enfermedad de Chagas se
encuentra únicamente en los países tropicales y
subtropicales de América Central y de Sudamérica. Los tripomastigotes
son más frágiles que los que causan la tripanosomiasis africana, se recomienda
la serología en lugar de la morfología para detectarlos iniciamlente, en el
laboratorio las pruebas que detectan los organismos móviles son más sensibles
que las que necesitan preparaciones fijadas y teñidas.

Figura. 134. T. cruzi. Muestra: Sangre
periférica.
Tinción: Giemsa. Aumento 1000x
Fuente: Lewis, S. J, B. Bates, Imelda. (2008).
Leishmaniasis.
Leishmaniasis es un grupo
de enfermedades parasitarias zoonóticas, producidas por diferentes especies de
protozoarios hemoflagelados del género Leishmania. Puede darse de forma:
Cutánea (afecta a la piel) Visceral (afecta a los órganos) Mucosa
(naso-orofaringea)
Agente etiológico.-
Protozoario dimórfico del género Leishmania, que pertenece al reino Protista,
subreino Protozoa, orden Kinetoplastida y a la familia Trypanosomatidae En la
actualidad, el género Leishmania se divide en dos subgéneros, según su
desarrollo en el intestino de los flebótomos vectores: Leishmania, en el
intestino medio o anterior, y Viannia, en el intestino posterior, medio y
anterior de los flebótomos. Morfológicamente las distintas especies de
leishmania no se pueden identificar. Para llegar a la clasificación de las
especies del género leishmania se debe considerar caracteristicas biologicas,
bioquimicas e inmunológicas.

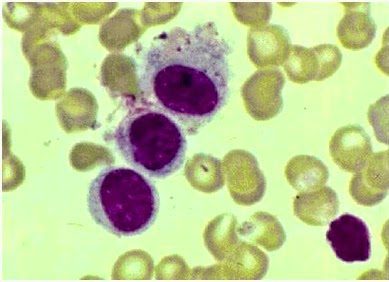
Figura 135. Parásitos de leishmania sp en
histiocitos.
Muestra. M.O. Aumento 1000x
Tinción: Wright
Fuente: (Ochoa, et al. 2009)
Referencias
bibliográficas.
Ochoa,
W. Gutiérrez, L. Guevara, R. Oviedo, M.
Loaiza, L. Bastidas, G. (2009).Diagnóstico de leishmaniosis visceral por frotis
de sangre periférica. A propósito de un caso en Cojedes, Venezuela. Recuperdo
de: http://www.scielo.org.pe/scielo.php?pid=S1726-46342009000200021&script=sci_arttext
Rodak B. (2004).
Hematología: fundamentos y aplicaciones clínicas. 2 ed. Rondinone S, trad.
Buenos Aires: Médica Panamericana.
Pérez, K. (s.f). Leishmaniasis. Recuperado de:
http://slideplayer.es/slide/121209/
Malaria o Paludismo (s.f.). viajarsano.com.
Recuperado de: https://barcelonaprosics.files.wordpress.com/2012/04/paludismo-viajarsano.pdf
Plasmodium falciparum (s.f). Malaria.
Recuperado de: http://mattressessale.eu/tag/plasmodium-falciparum
P.vivax. (2012, Junio 21). Microbiología
y Parasitología. Recuperado de:
http://2fmicrobiologia.blogspot.com/2012/06/plasmodium-vivax.html
Malaria (s.f). GEFOR. Recuperado de: http://www.gefor.4t.com/parasitologia/plasmodiumfalciparum.html
Lewis, S. J, B. Bates,
Imelda. (2008).Hematología práctica. Elsevier España Recuperdo de:
http://books.google.com.gt/books?id=6ka6vwRbogEC&dq=hematologia&q=chagas#v=snippet&q=chagas&f=false